Case study: command-line interface (CLI) tutorial
Nuno Saraiva-Agostinho
2026-03-09
Source:vignettes/CLI_tutorial.Rmd
CLI_tutorial.Rmdpsichomics is an interactive R package for integrative analyses of alternative splicing and gene expression based on The Cancer Genome Atlas (TCGA) (containing molecular data associated with 34 tumour types), the Genotype-Tissue Expression (GTEx) project (containing data for multiple normal human tissues), Sequence Read Archive (SRA) and user-provided data. The data from GTEx, TCGA and select SRA projects include subject/sample-associated information and transcriptomic data, such as the quantification of RNA-Seq reads aligning to splice junctions (henceforth called junction quantification) and exons.
Installing and starting the program
Install psichomics by typing the following in an R console (the R environment is required):
install.packages("BiocManager")
BiocManager::install("psichomics")After the installation, load psichomics by typing:
Quick reference of psichomics functions
Please read the following function reference.
Exploration of clinically-relevant, differentially spliced events in breast cancer
The following case study was adapted from psichomics’ original article:
Nuno Saraiva-Agostinho and Nuno L. Barbosa-Morais (2019). psichomics: graphical application for alternative splicing quantification and analysis. Nucleic Acids Research.
Breast cancer is the cancer type with the highest incidence and mortality in women (Torre et al., 2015) and multiple studies have suggested that transcriptome-wide analyses of alternative splicing changes in breast tumours are able to uncover tumour-specific biomarkers (Tsai et al., 2015; Danan-Gotthold et al., 2015; Anczuków et al., 2015). Given the relevance of early detection of breast cancer to patient survival, we can use psichomics to identify novel tumour stage-I-specific molecular signatures based on differentially spliced events.
Downloading and loading TCGA data
The quantification of each alternative splicing event is based on the proportion of junction reads that support the inclusion isoform, known as percent spliced-in or PSI (Wang et al., 2008).
To estimate this value for each splicing event, both alternative splicing annotation and junction quantification are required. While alternative splicing annotation is provided by the package, junction quantification may be retrieved from TCGA, GTEx, SRA or user-provided files.
Data is downloaded from FireBrowse, a service that hosts processed data from TCGA, as required to run the downstream analyses. Before downloading data, check the following options:
# Available tumour types
cohorts <- getFirebrowseCohorts()
# Available sample dates
date <- getFirebrowseDates()
# Available data types
dataTypes <- getFirebrowseDataTypes()Note there is also the option for Gene expression (normalised by RSEM). However, we recommend to load the raw gene expression data instead, followed by filtering and normalisation as demonstrated afterwards.
After deciding on the options to use, download and load breast cancer data as follows:
# Set download folder
folder <- getDownloadsFolder()
# Download and load most recent junction quantification and clinical data from
# TCGA/FireBrowse for Breast Cancer
data <- loadFirebrowseData(folder=folder,
cohort="BRCA",
data=c("clinical", "junction_quantification",
"RSEM_genes"),
date="2016-01-28")
names(data)
names(data[[1]])
# Select clinical and junction quantification dataset
clinical <- data[[1]]$`Clinical data`
sampleInfo <- data[[1]]$`Sample metadata`
junctionQuant <- data[[1]]$`Junction quantification (Illumina HiSeq)`
geneExpr <- data[[1]]$`Gene expression`Data is only downloaded if the files are not present in the given folder. In other words, if the files were already downloaded, the function will just load the files, so it is possible to reuse the code above just to load the requested files.
Windows limitations: If you are using Windows, note that the downloaded files have huge names that may be over Windows Maximum Path Length. A workaround would be to manually rename the downloaded files to have shorter names, move all downloaded files to a single folder and load such folder.
Filtering and normalising gene expression
As this package does not focuses on gene expression analysis, we
suggest to read the RNA-seq section of limma’s user guide.
Nevertheless, we present the following commands to quickly filter and
normalise gene expression:
# Check genes with 10 or more counts in at least some samples and 15 or more
# total counts across all samples
filter <- filterGeneExpr(geneExpr, minCounts=10, minTotalCounts=15)
# What normaliseGeneExpression() does:
# 1) Filter gene expression based on its argument "geneFilter"
# 2) Normalise gene expression with edgeR::calcNormFactors (internally) using
# the trimmed mean of M-values (TMM) method (by default)
# 3) Calculate log2-counts per million (logCPM)
geneExprNorm <- normaliseGeneExpression(geneExpr,
geneFilter=filter,
method="TMM",
log2transform=TRUE)Quantifying alternative splicing
After loading the clinical and alternative splicing junction quantification data from TCGA, quantify alternative splicing by clicking the green panel Alternative splicing quantification.
As previously mentioned, alternative splicing is quantified from the previously loaded junction quantification and an alternative splicing annotation file. To check current annotation files available:
# Available alternative splicing annotation
annotList <- listSplicingAnnotations()
annotList## Human hg19 (2016-10-11)
## "AH51461"
## Human hg19 (2017-10-20)
## "AH60272"
## Human hg38 (2018-04-30)
## "AH63657"
## Human hg19 from VAST-TOOLS (2021-06-15)
## "AH95569"
## Human hg38 from VAST-TOOLS (2021-06-15)
## "AH95570"
## Mus musculus mm9 from VAST-TOOLS (2021-06-15)
## "AH95571"
## Mus musculus mm10 from VAST-TOOLS (2021-06-15)
## "AH95572"
## Bos taurus bosTau6 from VAST-TOOLS (2021-06-15)
## "AH95573"
## Gallus gallus galGal3 from VAST-TOOLS (2021-06-15)
## "AH95574"
## Gallus gallus galGal4 from VAST-TOOLS (2021-06-15)
## "AH95575"
## Xenopus tropicalis xenTro3 from VAST-TOOLS (2021-06-15)
## "AH95576"
## Danio rerio danRer10 from VAST-TOOLS (2021-06-15)
## "AH95577"
## Branchiostoma lanceolatum braLan2 from VAST-TOOLS (2021-06-15)
## "AH95578"
## Strongylocentrotus purpuratus strPur4 from VAST-TOOLS (2021-06-15)
## "AH95579"
## Drosophila melanogaster dm6 from VAST-TOOLS (2021-06-15)
## "AH95580"
## Strigamia maritima strMar1 from VAST-TOOLS (2021-06-15)
## "AH95581"
## Caenorhabditis elegans ce11 from VAST-TOOLS (2021-06-15)
## "AH95582"
## Schmidtea mediterranea schMed31 from VAST-TOOLS (2021-06-15)
## "AH95583"
## Nematostella vectensis nemVec1 from VAST-TOOLS (2021-06-15)
## "AH95584"
## Arabidopsis thaliana araTha10 from VAST-TOOLS (2021-06-15)
## "AH95585"Custom splicing annotation: Additional alternative splicing annotations can be prepared for psichomics by parsing the annotation from programs like VAST-TOOLS, MISO, SUPPA and rMATS. Note that SUPPA and rMATS are able to create their splicing annotation based on transcript annotation. Please read Preparing alternative splicing annotations.
To quantify alternative splicing, first select the junction quantification, alternative splicing annotation and alternative splicing event type(s) of interest:
# Load Human (hg19/GRCh37 assembly) annotation
hg19 <- listSplicingAnnotations(assembly="hg19")[[1]]
annotation <- loadAnnotation(hg19)
# Available alternative splicing event types (skipped exon, alternative
# first/last exon, mutually exclusive exons, etc.)
getSplicingEventTypes()## Skipped exon
## "SE"
## Mutually exclusive exon
## "MXE"
## Alternative 5' splice site
## "A5SS"
## Alternative 3' splice site
## "A3SS"
## Alternative first exon
## "AFE"
## Alternative last exon
## "ALE"
## Alternative first exon (exon-centred - less reliable)
## "AFE_exon"
## Alternative last exon (exon-centred - less reliable)
## "ALE_exon"Afterwards, quantify alternative splicing using the previously defined parameters:
# Discard alternative splicing quantified using few reads
minReads <- 10 # default
psi <- quantifySplicing(annotation, junctionQuant, minReads=minReads)
# Check the identifier of the splicing events in the resulting table
events <- rownames(psi)
head(events)## [1] "SE_3_+_13661331_13663275_13663415_13667945_FBLN2"
## [2] "SE_3_+_57908750_57911572_57911661_57913023_SLMAP"
## [3] "ALE_3_+_57908750_57911572_57913023_SLMAP"
## [4] "SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2"
## [5] "SE_12_-_56558432_56558152_56558087_56557549_SMARCC2"
## [6] "AFE_4_+_56755098_56750094_56756389_EXOC1"Note that the event identifier (for instance,
SE_1_-_2125078_2124414_2124284_2121220_C1orf86) is composed
of:
- Event type (
SEstands for skipped exon) - Chromosome (
1) - Strand (
-) - Relevant coordinates depending on event type (in this case, the first constitutive exon’s end, the alternative exon’ start and end and the second constitutive exon’s start)
- Associated gene (
C1orf86)
Warning: all examples shown in this case study are performed using a small, yet representative subset of the available data. Therefore, values shown here may correspond to those when performing the whole analysis.
Data grouping
Let us create groups based on available samples types (i.e. Metastatic, Primary solid Tumor and Solid Tissue Normal) and tumour stages. As tumour stages are divided by sub-stages, we will merge sub-stages so as to have only tumour samples from stages I, II, III and IV (stage X samples are discarded as they are uncharacterised tumour samples).
# Group by normal and tumour samples
types <- createGroupByAttribute("Sample types", sampleInfo)
normal <- types$`Solid Tissue Normal`
tumour <- types$`Primary solid Tumor`
# Group by tumour stage (I, II, III or IV) or normal samples
stages <- createGroupByAttribute(
"patient.stage_event.pathologic_stage_tumor_stage", clinical)
groups <- list()
for (i in c("i", "ii", "iii", "iv")) {
stage <- Reduce(union,
stages[grep(sprintf("stage %s[a|b|c]{0,1}$", i), names(stages))])
# Include only tumour samples
stageTumour <- names(getSubjectFromSample(tumour, stage))
elem <- list(stageTumour)
names(elem) <- paste("Tumour Stage", toupper(i))
groups <- c(groups, elem)
}
groups <- c(groups, Normal=list(normal))
# Prepare group colours (for consistency across downstream analyses)
colours <- c("#6D1F95", "#FF152C", "#00C7BA", "#FF964F", "#00C65A")
names(colours) <- names(groups)
attr(groups, "Colour") <- colours
# Prepare normal versus tumour stage I samples
normalVSstage1Tumour <- groups[c("Tumour Stage I", "Normal")]
attr(normalVSstage1Tumour, "Colour") <- attr(groups, "Colour")
# Prepare normal versus tumour samples
normalVStumour <- list(Normal=normal, Tumour=tumour)
attr(normalVStumour, "Colour") <- c(Normal="#00C65A", Tumour="#EFE35C")Principal component analysis (PCA)
PCA is a technique to reduce data dimensionality by identifying variable combinations (called principal components) that explain the variance in the data (Ringnér, 2008). Use the following commands to perform PCA:
# PCA of PSI between normal and tumour stage I samples
psi_stage1Norm <- psi[ , unlist(normalVSstage1Tumour)]
pcaPSI_stage1Norm <- performPCA(t(psi_stage1Norm))As PCA cannot be performed on data with missing values, missing values need to be either removed (thus discarding data from whole splicing events or genes) or impute them (i.e. attributing to missing values the median of the non-missing ones). Use the argument
missingValuesofperformPCA()to select the number of missing values that are tolerable per event (i.e. if a splicing event or gene has less than N missing values, those missing values will be imputed; otherwise, the event is discarded from PCA).
# Explained variance across principal components
plotPCAvariance(pcaPSI_stage1Norm)
# Score plot (clinical individuals)
plotPCA(pcaPSI_stage1Norm, groups=normalVSstage1Tumour)
# Loading plot (variable contributions)
plotPCA(pcaPSI_stage1Norm, loadings=TRUE, individuals=FALSE,
nLoadings=100)For performance reasons, the loading plot is able to limit the number of top variables that most contribute to the select principal components, as controlled by the argument
nLoadingsofplotPCA().
Hint: As most plots in psichomics, PCA plots can be zoomed-in by clicking-and-dragging within the plot (click Reset zoom to zoom-out). To toggle the visibility of the data series represented in the plot, click its respective name in the plot legend.
# Table of variable contributions (as used to plot PCA, also)
table <- calculateLoadingsContribution(pcaPSI_stage1Norm)
head(table, 5)## Rank
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 1
## AFE_15_+_74466994_74466360_74467192_ISLR 2
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 3
## ALE_3_+_57908750_57911572_57913023_SLMAP 4
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 5
## Event type
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 Skipped exon (SE)
## AFE_15_+_74466994_74466360_74467192_ISLR Alternative first exon (AFE)
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP Skipped exon (SE)
## ALE_3_+_57908750_57911572_57913023_SLMAP Alternative last exon (ALE)
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 Skipped exon (SE)
## Chromosome Strand Gene
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 3 + FBLN2
## AFE_15_+_74466994_74466360_74467192_ISLR 15 + ISLR
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 3 + SLMAP
## ALE_3_+_57908750_57911572_57913023_SLMAP 3 + SLMAP
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 3 - LRRFIP2
## Event position
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 13661331, 13667945
## AFE_15_+_74466994_74466360_74467192_ISLR 74466360, 74467192
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 57908750, 57913023
## ALE_3_+_57908750_57911572_57913023_SLMAP 57908750, 57913023
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 37125297, 37136283
## PC1 loading PC2 loading
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 0.1339504 -0.14030197
## AFE_15_+_74466994_74466360_74467192_ISLR 0.1190302 -0.21011081
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 0.1365527 -0.05918619
## ALE_3_+_57908750_57911572_57913023_SLMAP 0.1358264 -0.06086915
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 0.1320250 -0.01416603
## Contribution to PC1 (%)
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 1.794271
## AFE_15_+_74466994_74466360_74467192_ISLR 1.416820
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 1.864663
## ALE_3_+_57908750_57911572_57913023_SLMAP 1.844881
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 1.743061
## Contribution to PC2 (%)
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 1.96846433
## AFE_15_+_74466994_74466360_74467192_ISLR 4.41465532
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 0.35030055
## ALE_3_+_57908750_57911572_57913023_SLMAP 0.37050534
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 0.02006764
## Contribution to PC1 and PC2 (%)
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 1.814085
## AFE_15_+_74466994_74466360_74467192_ISLR 1.757812
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 1.692410
## ALE_3_+_57908750_57911572_57913023_SLMAP 1.677176
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 1.547077To perform PCA using alternative splicing quantification and gene expression data (both using all samples and only Tumour Stage I and Normal samples):
# PCA of PSI between all samples (coloured by tumour stage and normal samples)
pcaPSI_all <- performPCA(t(psi))
plotPCA(pcaPSI_all, groups=groups)
plotPCA(pcaPSI_all, loadings=TRUE, individuals=FALSE)
# PCA of gene expression between all samples (coloured by tumour stage and
# normal samples)
pcaGE_all <- performPCA(t(geneExprNorm))
plotPCA(pcaGE_all, groups=groups)
plotPCA(pcaGE_all, loadings=TRUE, individuals=FALSE)
# PCA of gene expression between normal and tumour stage I samples
ge_stage1Norm <- geneExprNorm[ , unlist(normalVSstage1Tumour)]
pcaGE_stage1Norm <- performPCA(t(ge_stage1Norm))
plotPCA(pcaGE_stage1Norm, groups=normalVSstage1Tumour)
plotPCA(pcaGE_stage1Norm, loadings=TRUE, individuals=FALSE)NUMB exon 12 inclusion and correlation with QKI gene expression
One of the splicing events that most contribute the separation between tumour stage I and normal samples is NUMB exon 12 inclusion, whose protein is crucial for cell differentiation as a key regulator of the Notch pathway. The RNA-binding protein QKI has been shown to repress NUMB exon 12 inclusion in lung cancer cells by competing with core splicing factor SF1 for binding to the branch-point sequence, thereby repressing the Notch signalling pathway, which results in decreased cancer cell proliferation (Zong et al., 2014).
Differential inclusion of NUMB exon 12
Let’s check whether a significant difference in NUMB exon 12 inclusion between tumour and normal TCGA breast samples. To do so:
## [1] "SE_14_-_73749067_73746132_73745989_73744001_NUMB"
NUMBskippedExon12 <- tmp[1]
# Plot the representation of NUMB exon 12 inclusion
plotSplicingEvent(NUMBskippedExon12)
# Plot its PSI distribution
plotDistribution(psi[NUMBskippedExon12, ], normalVStumour)Consistent with the cited article, NUMB exon 12 inclusion is significantly increased in cancer.
Also of interest:
- Hover each group in the plot to compare the respective number of samples, median and variance.
- To zoom in a specific region, click-and-drag in the region of interest.
- To hide or show groups, click on their name in the legend.
Correlation between NUMB exon 12 inclusion and QKI expression
To verify if NUMB exon 12 inclusion is correlated with QKI expression:
## [1] "QKI|9444"
QKI <- tmp[1] # "QKI|9444"
# Plot its gene expression distribution
plotDistribution(geneExprNorm[QKI, ], normalVStumour, psi=FALSE)
plotCorrelation(correlateGEandAS(
geneExprNorm, psi, QKI, NUMBskippedExon12, method="spearman"))## $`SE_14_-_73749067_73746132_73745989_73744001_NUMB`
## $`SE_14_-_73749067_73746132_73745989_73744001_NUMB`$`QKI|9444`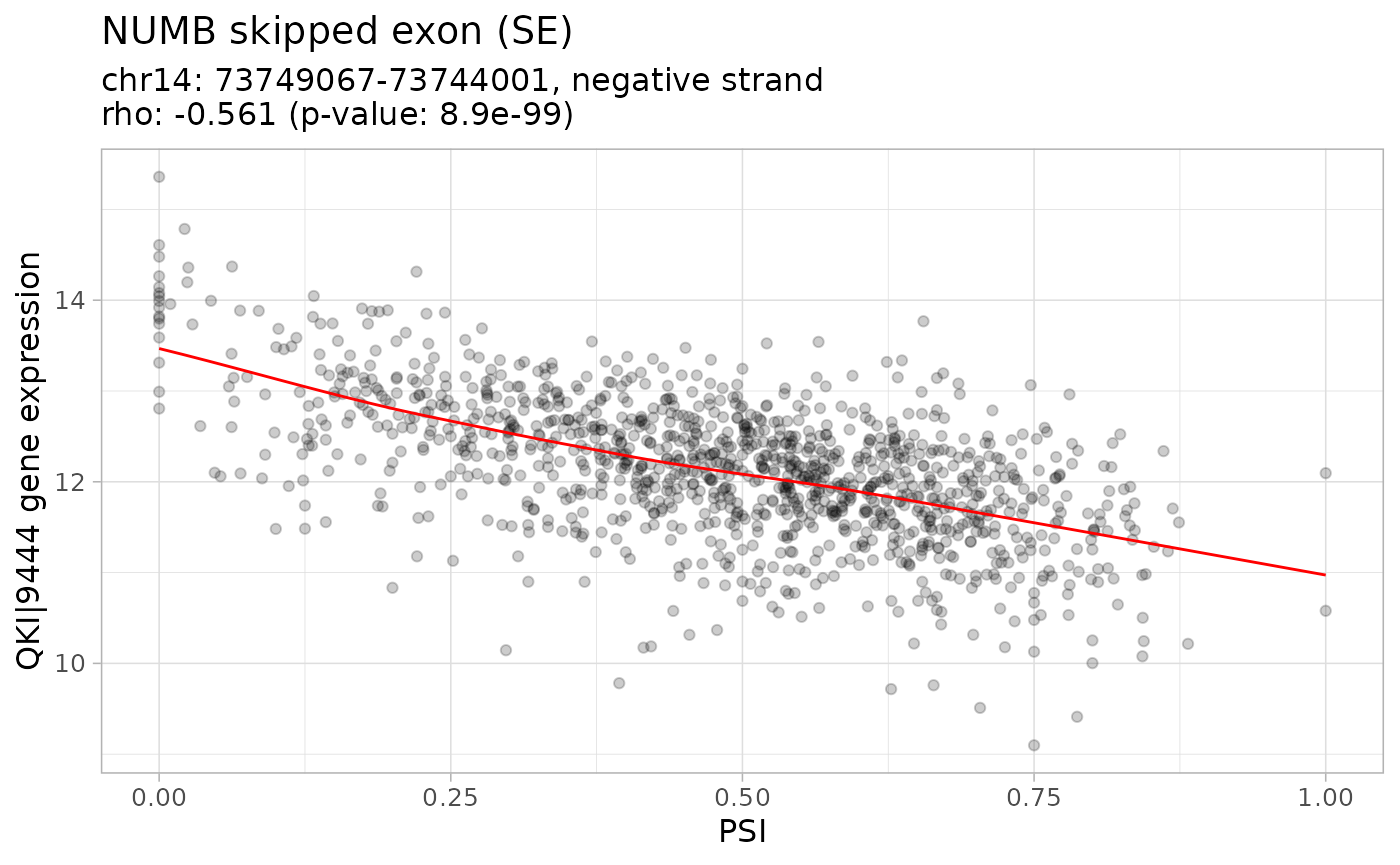
According to the obtained results and also consistent with the previous article, the inclusion of the exon is negatively correlated with QKI expression.
Differential splicing analysis
To analyse alternative splicing between normal and tumour stage I samples:
diffSplicing <- diffAnalyses(psi, normalVSstage1Tumour)
# Filter based on |∆ Median PSI| > 0.1 and q-value < 0.01
deltaPSIthreshold <- abs(diffSplicing$`∆ Median`) > 0.1
pvalueThreshold <- diffSplicing$`Wilcoxon p-value (BH adjusted)` < 0.01
eventsThreshold <- diffSplicing[deltaPSIthreshold & pvalueThreshold, ]
# Plot results
library(ggplot2)
ggplot(diffSplicing, aes(`∆ Median`,
-log10(`Wilcoxon p-value (BH adjusted)`))) +
geom_point(data=eventsThreshold,
colour="orange", alpha=0.5, size=3) +
geom_point(data=diffSplicing[!deltaPSIthreshold | !pvalueThreshold, ],
colour="gray", alpha=0.5, size=3) +
theme_light(16) +
ylab("-log10(q-value)")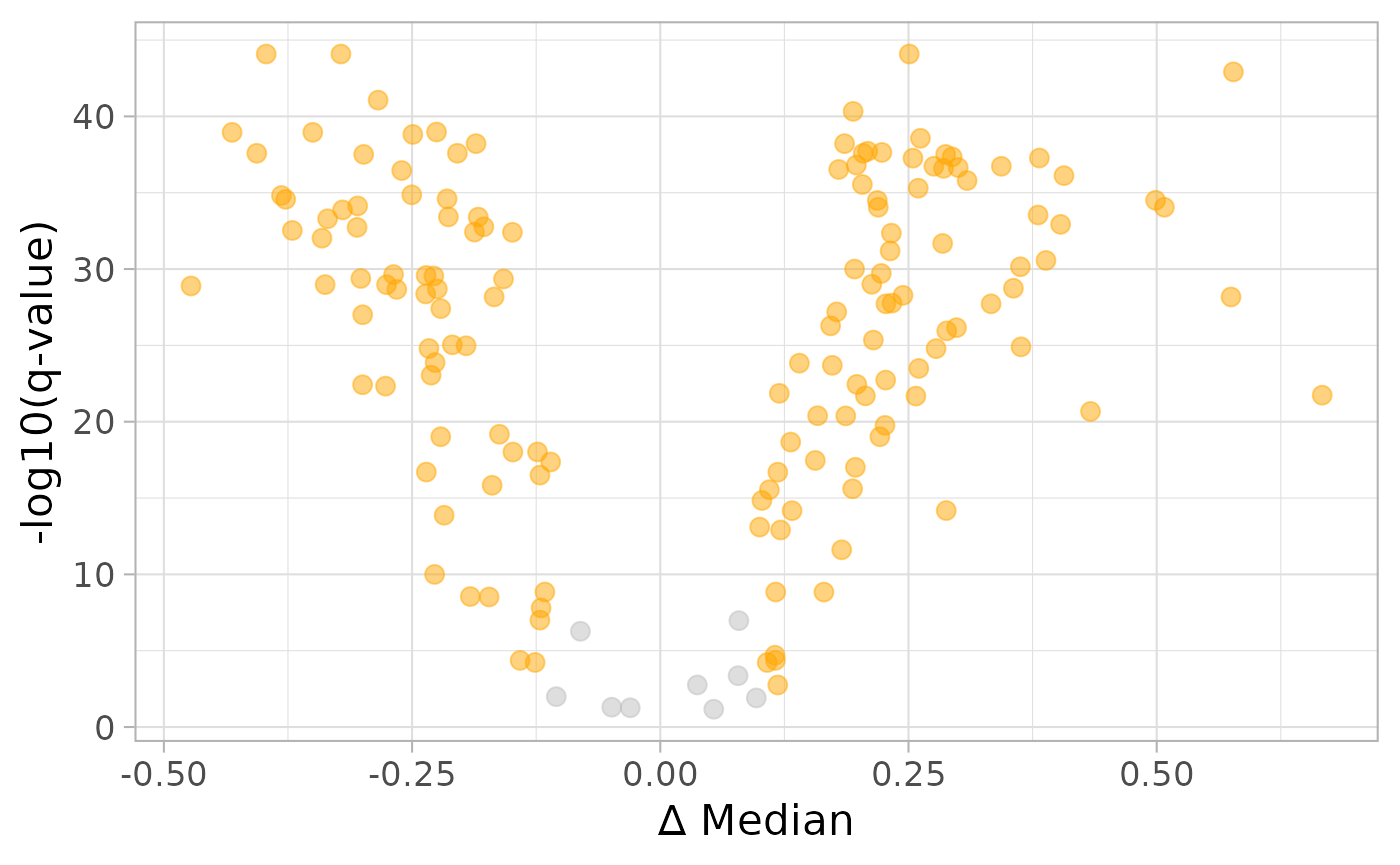
# Table of events that pass the thresholds
head(eventsThreshold)## Event type
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 Skipped exon (SE)
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP Skipped exon (SE)
## ALE_3_+_57908750_57911572_57913023_SLMAP Alternative last exon (ALE)
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 Skipped exon (SE)
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 Skipped exon (SE)
## AFE_4_+_56755098_56750094_56756389_EXOC1 Alternative first exon (AFE)
## Chromosome Strand Gene
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 3 + FBLN2
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 3 + SLMAP
## ALE_3_+_57908750_57911572_57913023_SLMAP 3 + SLMAP
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 3 - LRRFIP2
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 12 - SMARCC2
## AFE_4_+_56755098_56750094_56756389_EXOC1 4 + EXOC1
## Survival by PSI cutoff
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 <NA>
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP <NA>
## ALE_3_+_57908750_57911572_57913023_SLMAP <NA>
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 <NA>
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 <NA>
## AFE_4_+_56755098_56750094_56756389_EXOC1 <NA>
## Optimal PSI cutoff
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 <NA>
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP <NA>
## ALE_3_+_57908750_57911572_57913023_SLMAP <NA>
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 <NA>
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 <NA>
## AFE_4_+_56755098_56750094_56756389_EXOC1 <NA>
## Log-rank p-value
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 <NA>
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP <NA>
## ALE_3_+_57908750_57911572_57913023_SLMAP <NA>
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 <NA>
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 <NA>
## AFE_4_+_56755098_56750094_56756389_EXOC1 <NA>
## Samples (Normal)
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 112
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 112
## ALE_3_+_57908750_57911572_57913023_SLMAP 112
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 112
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 112
## AFE_4_+_56755098_56750094_56756389_EXOC1 112
## Samples (Tumour Stage I)
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 178
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 181
## ALE_3_+_57908750_57911572_57913023_SLMAP 181
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 180
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 181
## AFE_4_+_56755098_56750094_56756389_EXOC1 175
## T-test statistic
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 35.25121
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 22.08735
## ALE_3_+_57908750_57911572_57913023_SLMAP 21.76346
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 20.17074
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 -20.73860
## AFE_4_+_56755098_56750094_56756389_EXOC1 14.23168
## T-test parameter
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 256.8713
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 290.9932
## ALE_3_+_57908750_57911572_57913023_SLMAP 289.9965
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 251.8459
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 177.9125
## AFE_4_+_56755098_56750094_56756389_EXOC1 208.5711
## T-test p-value
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 2.113732e-100
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 3.636287e-64
## ALE_3_+_57908750_57911572_57913023_SLMAP 6.333372e-63
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 1.677488e-54
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 2.369187e-49
## AFE_4_+_56755098_56750094_56756389_EXOC1 1.444998e-32
## T-test p-value (BH adjusted)
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 3.276285e-98
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 9.393740e-63
## ALE_3_+_57908750_57911572_57913023_SLMAP 1.402389e-61
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 2.166756e-53
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 1.748686e-48
## AFE_4_+_56755098_56750094_56756389_EXOC1 3.199639e-32
## T-test conf int1
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 0.4913165
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 0.3970111
## ALE_3_+_57908750_57911572_57913023_SLMAP 0.3947718
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 0.3578812
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 -0.4497058
## AFE_4_+_56755098_56750094_56756389_EXOC1 0.2866482
## T-test conf int2
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 0.5494573
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 0.4746859
## ALE_3_+_57908750_57911572_57913023_SLMAP 0.4732735
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 0.4353285
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 -0.3715582
## AFE_4_+_56755098_56750094_56756389_EXOC1 0.3788319
## T-test estimate1
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 0.6951339
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 0.7505524
## ALE_3_+_57908750_57911572_57913023_SLMAP 0.7172262
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 0.7089629
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 0.3877788
## AFE_4_+_56755098_56750094_56756389_EXOC1 0.6677512
## T-test estimate2
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 0.1747470
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 0.3147039
## ALE_3_+_57908750_57911572_57913023_SLMAP 0.2832036
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 0.3123581
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 0.7984108
## AFE_4_+_56755098_56750094_56756389_EXOC1 0.3350112
## T-test null value
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 0
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 0
## ALE_3_+_57908750_57911572_57913023_SLMAP 0
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 0
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 0
## AFE_4_+_56755098_56750094_56756389_EXOC1 0
## T-test stderr
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 0.01476224
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 0.01973294
## ALE_3_+_57908750_57911572_57913023_SLMAP 0.01994273
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 0.01966239
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 0.01980038
## AFE_4_+_56755098_56750094_56756389_EXOC1 0.02338024
## T-test alternative
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 two.sided
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP two.sided
## ALE_3_+_57908750_57911572_57913023_SLMAP two.sided
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 two.sided
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 two.sided
## AFE_4_+_56755098_56750094_56756389_EXOC1 two.sided
## T-test method
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 Welch Two Sample t-test
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP Welch Two Sample t-test
## ALE_3_+_57908750_57911572_57913023_SLMAP Welch Two Sample t-test
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 Welch Two Sample t-test
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 Welch Two Sample t-test
## AFE_4_+_56755098_56750094_56756389_EXOC1 Welch Two Sample t-test
## T-test data name
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 vector[typeOne] and vector[!typeOne]
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP vector[typeOne] and vector[!typeOne]
## ALE_3_+_57908750_57911572_57913023_SLMAP vector[typeOne] and vector[!typeOne]
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 vector[typeOne] and vector[!typeOne]
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 vector[typeOne] and vector[!typeOne]
## AFE_4_+_56755098_56750094_56756389_EXOC1 vector[typeOne] and vector[!typeOne]
## Wilcoxon statistic
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 19781.5
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 18879.0
## ALE_3_+_57908750_57911572_57913023_SLMAP 18942.5
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 19065.0
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 694.0
## AFE_4_+_56755098_56750094_56756389_EXOC1 17428.0
## Wilcoxon p-value
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 3.138055e-45
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 2.447864e-35
## ALE_3_+_57908750_57911572_57913023_SLMAP 7.906559e-36
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 1.521918e-37
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 6.272224e-41
## AFE_4_+_56755098_56750094_56756389_EXOC1 9.851691e-29
## Wilcoxon p-value (BH adjusted)
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 1.215996e-43
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 8.983718e-35
## ALE_3_+_57908750_57911572_57913023_SLMAP 3.142350e-35
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 7.609591e-37
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 1.111826e-39
## AFE_4_+_56755098_56750094_56756389_EXOC1 1.896335e-28
## Wilcoxon null value
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 0
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 0
## ALE_3_+_57908750_57911572_57913023_SLMAP 0
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 0
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 0
## AFE_4_+_56755098_56750094_56756389_EXOC1 0
## Wilcoxon alternative
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 two.sided
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP two.sided
## ALE_3_+_57908750_57911572_57913023_SLMAP two.sided
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 two.sided
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 two.sided
## AFE_4_+_56755098_56750094_56756389_EXOC1 two.sided
## Wilcoxon method
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 Wilcoxon rank sum test with continuity correction
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP Wilcoxon rank sum test with continuity correction
## ALE_3_+_57908750_57911572_57913023_SLMAP Wilcoxon rank sum test with continuity correction
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 Wilcoxon rank sum test with continuity correction
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 Wilcoxon rank sum test with continuity correction
## AFE_4_+_56755098_56750094_56756389_EXOC1 Wilcoxon rank sum test with continuity correction
## Wilcoxon data name
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 vector[typeOne] and vector[!typeOne]
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP vector[typeOne] and vector[!typeOne]
## ALE_3_+_57908750_57911572_57913023_SLMAP vector[typeOne] and vector[!typeOne]
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 vector[typeOne] and vector[!typeOne]
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 vector[typeOne] and vector[!typeOne]
## AFE_4_+_56755098_56750094_56756389_EXOC1 vector[typeOne] and vector[!typeOne]
## Kruskal statistic
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 199.2100
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 153.9075
## ALE_3_+_57908750_57911572_57913023_SLMAP 156.1536
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 164.0062
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 179.5060
## AFE_4_+_56755098_56750094_56756389_EXOC1 123.7057
## Kruskal parameter
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 1
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 1
## ALE_3_+_57908750_57911572_57913023_SLMAP 1
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 1
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 1
## AFE_4_+_56755098_56750094_56756389_EXOC1 1
## Kruskal p-value
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 3.106209e-45
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 2.426277e-35
## ALE_3_+_57908750_57911572_57913023_SLMAP 7.836334e-36
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 1.508009e-37
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 6.212560e-41
## AFE_4_+_56755098_56750094_56756389_EXOC1 9.771502e-29
## Kruskal p-value (BH adjusted)
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 1.203656e-43
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 8.904501e-35
## ALE_3_+_57908750_57911572_57913023_SLMAP 3.114440e-35
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 7.540044e-37
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 1.101252e-39
## AFE_4_+_56755098_56750094_56756389_EXOC1 1.881109e-28
## Kruskal method
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 Kruskal-Wallis rank sum test
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP Kruskal-Wallis rank sum test
## ALE_3_+_57908750_57911572_57913023_SLMAP Kruskal-Wallis rank sum test
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 Kruskal-Wallis rank sum test
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 Kruskal-Wallis rank sum test
## AFE_4_+_56755098_56750094_56756389_EXOC1 Kruskal-Wallis rank sum test
## Kruskal data name
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 vector and group
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP vector and group
## ALE_3_+_57908750_57911572_57913023_SLMAP vector and group
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 vector and group
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 vector and group
## AFE_4_+_56755098_56750094_56756389_EXOC1 vector and group
## Levene statistic
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 0.003080886
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 10.444152452
## ALE_3_+_57908750_57911572_57913023_SLMAP 7.806604006
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 1.574398815
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 27.572904067
## AFE_4_+_56755098_56750094_56756389_EXOC1 3.337182318
## Levene p-value
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 9.557741e-01
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 1.371647e-03
## ALE_3_+_57908750_57911572_57913023_SLMAP 5.551130e-03
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 2.105796e-01
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 2.924722e-07
## AFE_4_+_56755098_56750094_56756389_EXOC1 6.877635e-02
## Levene p-value (BH adjusted)
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 9.557741e-01
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 2.472154e-03
## ALE_3_+_57908750_57911572_57913023_SLMAP 9.057107e-03
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 2.417766e-01
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 8.242399e-07
## AFE_4_+_56755098_56750094_56756389_EXOC1 8.810194e-02
## Levene data name
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 vector and group
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP vector and group
## ALE_3_+_57908750_57911572_57913023_SLMAP vector and group
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 vector and group
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 vector and group
## AFE_4_+_56755098_56750094_56756389_EXOC1 vector and group
## Levene method
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 Levene's test (using the median)
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP Levene's test (using the median)
## ALE_3_+_57908750_57911572_57913023_SLMAP Levene's test (using the median)
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 Levene's test (using the median)
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 Levene's test (using the median)
## AFE_4_+_56755098_56750094_56756389_EXOC1 Levene's test (using the median)
## Fligner-Killeen statistic
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 0.5378107
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 7.0960805
## ALE_3_+_57908750_57911572_57913023_SLMAP 5.3586469
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 1.6230249
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 23.2937373
## AFE_4_+_56755098_56750094_56756389_EXOC1 3.2181098
## Fligner-Killeen parameter
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 1
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 1
## ALE_3_+_57908750_57911572_57913023_SLMAP 1
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 1
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 1
## AFE_4_+_56755098_56750094_56756389_EXOC1 1
## Fligner-Killeen p-value
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 4.633415e-01
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 7.725270e-03
## ALE_3_+_57908750_57911572_57913023_SLMAP 2.061977e-02
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 2.026705e-01
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 1.390520e-06
## AFE_4_+_56755098_56750094_56756389_EXOC1 7.282768e-02
## Fligner-Killeen p-value (BH adjusted)
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 5.022233e-01
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 1.221854e-02
## ALE_3_+_57908750_57911572_57913023_SLMAP 2.853628e-02
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 2.344323e-01
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 3.653062e-06
## AFE_4_+_56755098_56750094_56756389_EXOC1 9.329166e-02
## Fligner-Killeen method
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 Fligner-Killeen test of homogeneity of variances
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP Fligner-Killeen test of homogeneity of variances
## ALE_3_+_57908750_57911572_57913023_SLMAP Fligner-Killeen test of homogeneity of variances
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 Fligner-Killeen test of homogeneity of variances
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 Fligner-Killeen test of homogeneity of variances
## AFE_4_+_56755098_56750094_56756389_EXOC1 Fligner-Killeen test of homogeneity of variances
## Fligner-Killeen data name
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 vector and group
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP vector and group
## ALE_3_+_57908750_57911572_57913023_SLMAP vector and group
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 vector and group
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 vector and group
## AFE_4_+_56755098_56750094_56756389_EXOC1 vector and group
## Variance (Normal)
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 0.01354229
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 0.01673786
## ALE_3_+_57908750_57911572_57913023_SLMAP 0.01826371
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 0.02476543
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 0.03375412
## AFE_4_+_56755098_56750094_56756389_EXOC1 0.04191694
## Variance (Tumour Stage I)
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 0.01726787
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 0.04342983
## ALE_3_+_57908750_57911572_57913023_SLMAP 0.04247048
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 0.02978812
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 0.01641286
## AFE_4_+_56755098_56750094_56756389_EXOC1 0.03016604
## Median (Normal)
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 0.7189603
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 0.7547746
## ALE_3_+_57908750_57911572_57913023_SLMAP 0.7195130
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 0.6925958
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 0.3897698
## AFE_4_+_56755098_56750094_56756389_EXOC1 0.6466063
## Median (Tumour Stage I)
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 0.1416915
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP 0.2470588
## ALE_3_+_57908750_57911572_57913023_SLMAP 0.2206573
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 0.2860301
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 0.8210526
## AFE_4_+_56755098_56750094_56756389_EXOC1 0.3134328
## ∆ Variance ∆ Median
## SE_3_+_13661331_13663275_13663415_13667945_FBLN2 -0.003725583 0.5772688
## SE_3_+_57908750_57911572_57911661_57913023_SLMAP -0.026691961 0.5077157
## ALE_3_+_57908750_57911572_57913023_SLMAP -0.024206769 0.4988557
## SE_3_-_37136283_37133029_37132958_37125297_LRRFIP2 -0.005022695 0.4065657
## SE_12_-_56558432_56558152_56558087_56557549_SMARCC2 0.017341258 -0.4312828
## AFE_4_+_56755098_56750094_56756389_EXOC1 0.011750897 0.3331735Performing multiple survival analysis
To study the impact of alternative splicing events on prognosis, Kaplan-Meier curves may be plotted for groups of patients separated by the optimal PSI cutoff for a given alternative splicing event that that maximises the significance of group differences in survival analysis (i.e. minimises the p-value of the log-rank tests of difference in survival between individuals whose samples have their PSI below and above that threshold).
Given the slow process of calculating the optimal splicing quantification cutoff for multiple events, it is recommended to perform this for a subset of differentially spliced events.
# Events already tested which have prognostic value
events <- c(
"SE_9_+_6486925_6492303_6492401_6493826_UHRF2",
"SE_4_-_87028376_87024397_87024339_87023185_MAPK10",
"SE_2_+_152324660_152324988_152325065_152325155_RIF1",
"SE_2_+_228205096_228217230_228217289_228220393_MFF",
"MXE_15_+_63353138_63353397_63353472_63353912_63353987_63354414_TPM1",
"SE_2_+_173362828_173366500_173366629_173368819_ITGA6",
"SE_1_+_204957934_204971724_204971876_204978685_NFASC")
# Survival curves based on optimal PSI cutoff
library(survival)
# Assign alternative splicing quantification to patients based on their samples
samples <- colnames(psi)
match <- getSubjectFromSample(samples, clinical, sampleInfo=sampleInfo)
survPlots <- list()
for (event in events) {
# Find optimal cutoff for the event
eventPSI <- assignValuePerSubject(psi[event, ], match, clinical,
samples=unlist(tumour))
opt <- optimalSurvivalCutoff(clinical, eventPSI, censoring="right",
event="days_to_death",
timeStart="days_to_death")
(optimalCutoff <- opt$par) # Optimal exon inclusion level
(optimalPvalue <- opt$value) # Respective p-value
label <- labelBasedOnCutoff(eventPSI, round(optimalCutoff, 2),
label="PSI values")
survTerms <- processSurvTerms(clinical, censoring="right",
event="days_to_death",
timeStart="days_to_death",
group=label, scale="years")
surv <- survfit(survTerms)
pvalue <- testSurvival(survTerms)
survPlots[[event]] <- plotSurvivalCurves(surv, pvalue=pvalue, mark=FALSE)
}
# Now print the survival plot of a specific event
survPlots[[ events[[1]] ]]Differential gene expression
Detected alterations in alternative splicing may simply be a reflection of changes in gene expression levels. Therefore, to disentangle these two effects, differential expression analysis between tumour stage I and normal samples should also be performed. In order to do so:
# Prepare groups of samples to analyse and further filter unavailable samples in
# selected groups for gene expression
ge <- geneExprNorm[ , unlist(normalVSstage1Tumour), drop=FALSE]
isFromGroup1 <- colnames(ge) %in% normalVSstage1Tumour[[1]]
design <- cbind(1, ifelse(isFromGroup1, 0, 1))
# Fit a gene-wise linear model based on selected groups
library(limma)
fit <- lmFit(as.matrix(ge), design)
# Calculate moderated t-statistics and DE log-odds using limma::eBayes
ebayesFit <- eBayes(fit, trend=TRUE)
# Prepare data summary
pvalueAdjust <- "BH" # Benjamini-Hochberg p-value adjustment (FDR)
summary <- topTable(ebayesFit, number=nrow(fit), coef=2, sort.by="none",
adjust.method=pvalueAdjust, confint=TRUE)
names(summary) <- c("log2 Fold-Change", "CI (low)", "CI (high)",
"Average expression", "moderated t-statistics", "p-value",
paste0("p-value (", pvalueAdjust, " adjusted)"),
"B-statistics")
attr(summary, "groups") <- normalVSstage1Tumour
# Calculate basic statistics
stats <- diffAnalyses(ge, normalVSstage1Tumour, "basicStats",
pvalueAdjust=NULL)
final <- cbind(stats, summary)
# Differential gene expression between breast tumour stage I and normal samples
library(ggplot2)
library(ggrepel)
cognateGenes <- unlist(parseSplicingEvent(events)$gene)
logFCthreshold <- abs(final$`log2 Fold-Change`) > 1
pvalueThreshold <- final$`p-value (BH adjusted)` < 0.01
final$genes <- gsub("\\|.*$", "\\1", rownames(final))
ggplot(final, aes(`log2 Fold-Change`,
-log10(`p-value (BH adjusted)`))) +
geom_point(data=final[logFCthreshold & pvalueThreshold, ],
colour="orange", alpha=0.5, size=3) +
geom_point(data=final[!logFCthreshold | !pvalueThreshold, ],
colour="gray", alpha=0.5, size=3) +
geom_text_repel(data=final[cognateGenes, ], aes(label=genes),
box.padding=0.4, size=5) +
theme_light(16) +
ylab("-log10(q-value)")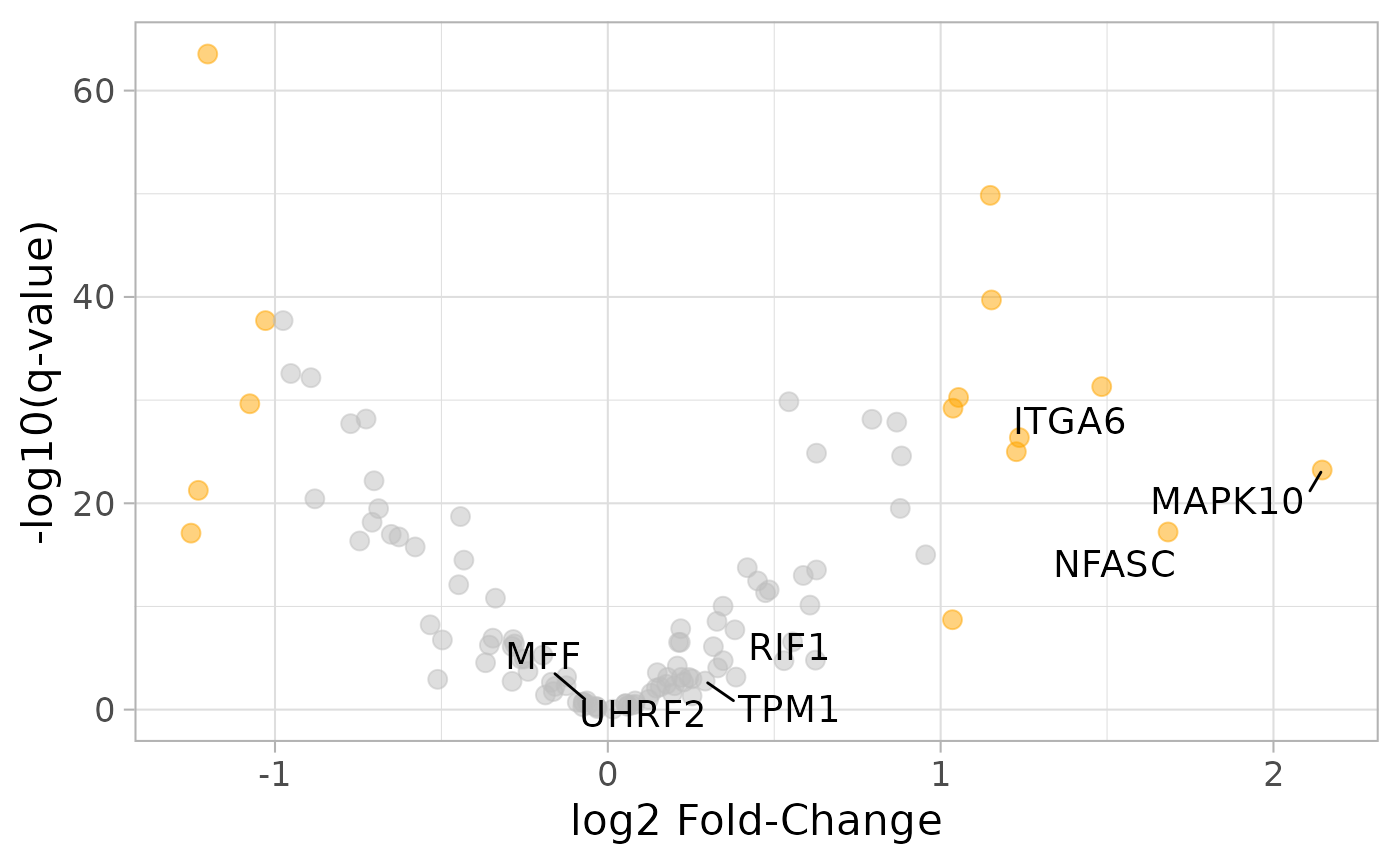
UHRF2 exon 10 inclusion
One splicing event with prognostic value is the alternative splicing of UHRF2 exon 10. Cell-cycle regulator UHRF2 promotes cell proliferation and inhibits the expression of tumour suppressors in breast cancer (Wu et al., 2012).
Differential splicing analysis
Let’s check whether a significant difference in UHRF2 exon 10 inclusion between tumour stage I and normal samples. To do so:
# UHRF2 skipped exon 10's PSI values per tumour stage I and normal samples
UHRF2skippedExon10 <- events[1]
plotDistribution(psi[UHRF2skippedExon10, ], normalVSstage1Tumour)Higher inclusion of UHRF2 exon 10 is associated with normal samples.
Survival analysis
To study the impact of alternative splicing events on prognosis, Kaplan-Meier curves may be plotted for groups of patients separated by a given PSI cutoff for a given alternative splicing event. The optimal PSI cutoff maximises the significance of group differences in survival analysis (i.e. minimises the p-value of the log-rank tests of difference in survival between individuals whose samples have a PSI below and above that threshold).
# Find optimal cutoff for the event
UHRF2skippedExon10 <- events[1]
eventPSI <- assignValuePerSubject(psi[UHRF2skippedExon10, ], match, clinical,
samples=unlist(tumour))
opt <- optimalSurvivalCutoff(clinical, eventPSI, censoring="right",
event="days_to_death", timeStart="days_to_death")
(optimalCutoff <- opt$par) # Optimal exon inclusion level## [1] 0.1436954
(optimalPvalue <- opt$value) # Respective p-value## [1] 0.0358
label <- labelBasedOnCutoff(eventPSI, round(optimalCutoff, 2),
label="PSI values")
survTerms <- processSurvTerms(clinical, censoring="right",
event="days_to_death", timeStart="days_to_death",
group=label, scale="years")
surv <- survfit(survTerms)
pvalue <- testSurvival(survTerms)
plotSurvivalCurves(surv, pvalue=pvalue, mark=FALSE)As per the results, higher inclusion of UHRF2 exon 10 is associated with better prognosis.
Differential expression
To check whether alternative splicing changes are related with gene expression alterations, let us perform differential expression analysis on UHRF2:
plotDistribution(geneExprNorm["UHRF2", ], normalVSstage1Tumour)It seems UHRF2 is differentially expressed between Tumour Stage I and Solid Tissue Normal. However, going back to exploratory differential gene expression, UHRF2 has a log2(fold-change) ≤ 1, low enough not to be biologically relevant. Following this criterium, the gene can thus be considered not to be differentially expressed between these conditions.
Survival analysis
To confirm if gene expression has an overall prognostic value, perform the following:
UHRF2ge <- assignValuePerSubject(geneExprNorm["UHRF2", ], match, clinical,
samples=unlist(tumour))
# Survival curves based on optimal gene expression cutoff
opt <- optimalSurvivalCutoff(clinical, UHRF2ge, censoring="right",
event="days_to_death", timeStart="days_to_death")
(optimalCutoff <- opt$par) # Optimal exon inclusion level## [1] 10.42619
(optimalPvalue <- opt$value) # Respective p-value## [1] 0.176
# Process again after rounding the cutoff
roundedCutoff <- round(optimalCutoff, 2)
label <- labelBasedOnCutoff(UHRF2ge, roundedCutoff, label="Gene expression")
survTerms <- processSurvTerms(clinical, censoring="right",
event="days_to_death", timeStart="days_to_death",
group=label, scale="years")
surv <- survfit(survTerms)
pvalue <- testSurvival(survTerms)
plotSurvivalCurves(surv, pvalue=pvalue, mark=FALSE)There seems to be no significant difference in survival between patient groups stratified by UHRF2’s optimal gene expression cutoff in tumour samples (log-rank p-value > 0.05).
Literature support and external database information
If an event is differentially spliced and has an impact on patient survival, its association with the studied disease might be already described in the literature. To check so, go to Analyses > Gene, transcript and protein information where information regarding the associated gene (such as description and genomic position), transcripts and protein domain annotation are available.
- The protein plot shows the UniProt matches for the selected transcript. Hover the protein’s rendered domains to obtain more information on them. More information about each protein can be retrieved by clicking the respective UniProt link.
- Links to related research articles are also available. Click Show more articles to be directed to PubMed.
- Multiple links to related external databases are available too:
- Human Protein Atlas (Cancer Atlas) allows to check the evidence of a gene at protein level for multiple cancer tissues.
- VastDB shows multi-species alternative splicing profiles for diverse tissues and cell types.
- UCSC Genome Browser may reveal protein domain disruptions caused by the alternative splicing event. To check so, activate the Pfam in UCSC Gene and UniProt tracks (in Genes and Gene Predictions) and check if any domains are annotated in the alternative and/or constitutive exons of the splicing event.
Interpretation
Higher inclusion of UHRF2 exon 10 is associated with normal samples and better prognosis, and potentially disrupts UHRF2’s SRA-YDG protein domain, related to the binding affinity to epigenetic marks. Hence, exon 10 inclusion may suppress UHRF2’s oncogenic role in breast cancer by impairing its activity through the induction of a truncated protein or a non-coding isoform. Moreover, this hypothesis is independent from gene expression changes, as UHRF2 is not differentially expressed between tumour stage I and normal samples (|log2(fold-change)| < 1) and there is no significant difference in survival between patient groups stratified by its expression in tumour samples (log-rank p-value > 0.05).
Loading data from other sources
Load GTEx data
GTEx data (subject phenotype, sample attributes, gene expression and junction quantification) for specific tissues can be automatically retrieved and loaded by following these commands:
# Check GTEx tissues available based on the sample attributes
getGtexTissues(sampleAttr)
tissues <- c("blood", "brain")
gtex <- loadGtexData("~/Downloads", tissues=tissues)
names(gtex)
names(gtex[[1]])To load data for all GTEx tissues, please type:
gtex <- loadGtexData("~/Downloads", tissues=NULL)
names(gtex)
names(gtex[[1]])Load SRA project data using recount
recount is a resource of pre-processed data for thousands of SRA projects (including gene read counts, splice junction quantification and sample metadata). psichomics supports automatic downloading and loading of SRA data from recount, as exemplified below:
View(recount::recount_abstract)
sra <- loadSRAproject("SRP053101")
names(sra)
names(sra[[1]])Please refer to our methods article for more information (the code for performing the analysis can be found at GitHub):
Nuno Saraiva-Agostinho and Nuno L. Barbosa-Morais (2020). Interactive Alternative Splicing Analysis of Human Stem Cells Using psichomics. In: Kidder B. (eds) Stem Cell Transcriptional Networks. Methods in Molecular Biology, vol 2117. Humana, New York, NY
Load user-provided data
Any FASTQ files can be manually aligned using a splice-aware aligner and loaded by following the instructions in Loading user-provided data.
Local files can be loaded by indicating their containing folder. Any files located in this folder and sub-folders will be loaded.
For instance, to load GTEx data via local files, create a directory called GTEx, put all GTEx files within that folder and type these commands:
folder <- "~/Downloads/GTEx/"
ignore <- c(".aux.", ".mage-tab.") # File patterns to ignore
data <- loadLocalFiles(folder, ignore=ignore)[[1]]
names(data)
names(data[[1]])
# Select clinical and junction quantification dataset
clinical <- data[["Clinical data"]]
sampleInfo <- data[["Sample metadata"]]
geneExpr <- data[["Gene expression"]]
junctionQuant <- data[["Junction quantification"]]Feedback
All feedback on the program, documentation and associated material (including this tutorial) is welcome. Please send any suggestions and comments to:
Nuno Saraiva-Agostinho (nunodanielagostinho@gmail.com)
Disease Transcriptomics Lab, Instituto de Medicina Molecular (Portugal)